
Computerunterstütztes Drug Design in der Arzneistoffentwicklung
26.01.2012 -
-
 Dr. Daniela Schuster, Institut für Pharmazie/Pharmazeutische Chemie, Universität Innsbruck, Österreich
Dr. Daniela Schuster, Institut für Pharmazie/Pharmazeutische Chemie, Universität Innsbruck, Österreich -
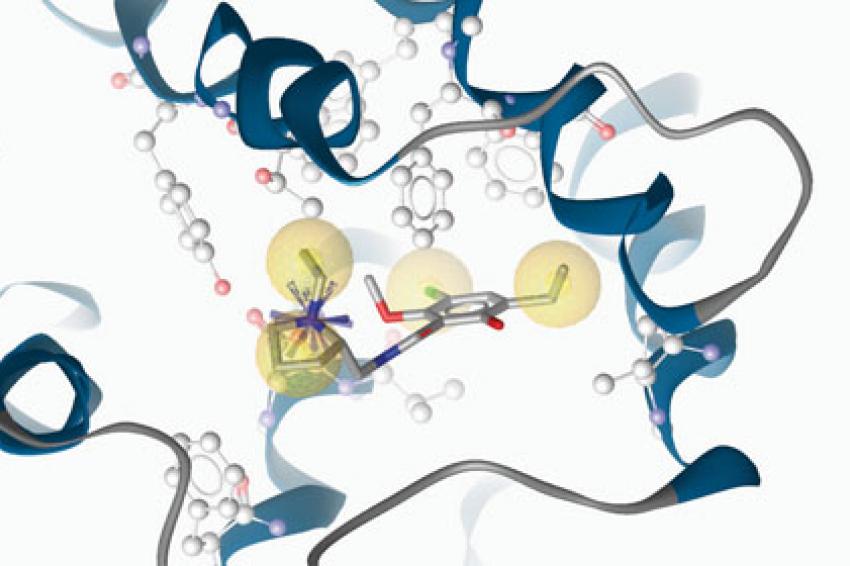 Abb. 1: Der Dopaminrezeptorantagonist Eticloprid in der Bindetasche des Dopamin-D3-Rezeptors (Koordinaten aus der Protein Data Bank, www.pdb.org, Eintrag 3PBL). Vollständige Legende siehe Haupttext
Abb. 1: Der Dopaminrezeptorantagonist Eticloprid in der Bindetasche des Dopamin-D3-Rezeptors (Koordinaten aus der Protein Data Bank, www.pdb.org, Eintrag 3PBL). Vollständige Legende siehe Haupttext -
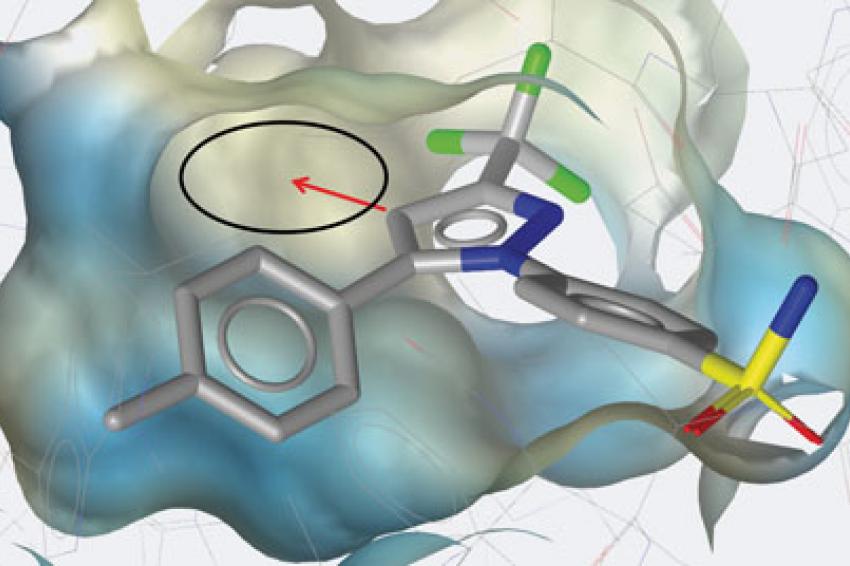 Abb. 2: Celecoxib in der Cyclooxygenase-2 Bindetasche (Protein Data Bank Eintrag 3LN1 dargestellt mit LigandScout). Vollständige Legende siehe Haupttext
Abb. 2: Celecoxib in der Cyclooxygenase-2 Bindetasche (Protein Data Bank Eintrag 3LN1 dargestellt mit LigandScout). Vollständige Legende siehe Haupttext
Ein vertieftes Verständnis für molekulare Wirkmechanismen, die Verfügbarkeit von Röntgenkristallstrukturen wichtiger Arzneitstofftargets und leistungsfähige Computer ermöglichen virtuelle Aktivitätsvorhersagen in der Entwicklung neuer Medikamente.
Computerbasierte Methoden haben in der Leitstruktursuche und -optimierung einen festen Platz. Sie können aber auch verwendet werden, um für einzelne Verbindungen ganze Aktivitätsprofile vorherzusagen und so zusätzliche Wirkmechanismen und Nebenwirkungen aufdecken. So leistet in silico Drug Design seinen Beitrag zu neuen, sicheren Arzneistoffen für die Zukunft.
Die heutige Arzneimittelforschung ist sehr aufwendig und häufig leider nur von moderaten Erfolgen gekrönt. So belaufen sich die Kosten, ein neues Medikament auf den Markt zu bringen, auf weit über eine Milliarde Dollar. Trotz steigender Ausgaben in der Arzneimittelforschung stagniert die Anzahl neu zugelassener, niedermolekularer Arzneistoffe in den letzten Jahren. Denn nicht nur die hohen Kosten, auch gestiegene Sicherheitsanforderungen der Zulassungsbehörden stellen die Entwickler neuer Medikamente vor große Herausforderungen.
Zu Beginn der Medikamentenentwicklung steht man vor der Frage, welche Verbindungen für weitere Studien überhaupt infrage kommen. Als eine mögliche Strategie zur Auswahl geeigneter Substanzen bietet sich computerunterstütztes Wirkstoffdesign an. Mithilfe von Computermodellen werden Moleküle vorgeschlagen, die eine gewünschte Wirkung zeigen könnten. Die biologische Austestung dieser Verbindungen führt in der Regel zu höheren Erfolgsraten als die Testung zufällig ausgewählter Substanzen und hilft so, den experimentellen Aufwand in der frühen Arzneimittelentwicklungs-Phase zu reduzieren. Diese Methoden kommen vor allem in der Leitstruktursuche und -optimierung zum Einsatz, wo sie sowohl auf synthetisch-organische Verbindungen als auch auf Naturstoffe angewandt werden.
Den Computermodellen liegen zwei einfache Prinzipien zugrunde:
- Ähnliche Moleküle haben ähnliche Bioaktivitäten (Liganden-basierter Ansatz) und
- Ein Molekül und sein Zielprotein müssen wie Schlüssel und Schloss zusammenpassen (Target-basierter Ansatz).
Je nach Verfahren werden „neue" Moleküle mit bereits bekannten aktiven verglichen (Abb. 1) oder der Testkandidat gleich direkt in die Protein-Bindetasche modelliert. Im zweiten Fall kann das Testmolekül auch so verändert werden, dass es die Bindetasche noch optimaler ausfüllt, wodurch man sich eine verbesserte Wirkstärke erwartet (Abb. 2). Zusammen mit Medizinalchemikern kann so die Leitstrukturoptimierung gezielt angegangen werden.
Abgesehen von der Leitstrukturfindung und -optimierung ist es auch möglich, bisher unbekannte Wirkungen bekannter Arzneistoffe zu identifizieren. So konnten in einer Studie der University of San Francisco für zehn Arzneistoffe aus unterschiedlichen Bereichen weitere, unerwartete Targets berechnet und experimentell bestätigt werden. Beispielsweise wurden das Neuroleptikum Fluanison als potenter α1-Rezeptorblocker und die Antidepressiva Fluoxetin und Paroxetin als Betablocker identifiziert. Diese Erkenntnisse können sowohl Nebenwirkungen erklären als auch potentielle neue Anwendungsgebiete dieser Arzneimittel aufzeigen.
Wenn man verschiedene Computermodelle gleichzeitig verwendet, die zahlreiche Zielproteine abdecken, kann man mit einem sogenannten parallelen Screening eine Verbindung auf ihr Aktivitätsprofil hin untersuchen. Auch hier müssen die vorgeschlagenen Wirkungen experimentell bestätigt werden. In einer Kollaboration österreichischer Universitäten wurde ein Aktivitätsprofil für Leoligin, ein Inhaltsstoff der Edelweißwurzel, erstellt. Als ein mögliches Target wurde das Cholesterylestertransferprotein vorgeschlagen, was in vitro und in vivo bestätigt werden konnte.
Computerunterstützte Methoden können auch helfen, dort biologische Wirkungen aufzuzeigen, wo man sie gar nicht haben will. So können etwa Umweltchemikalien, wie man sie in Badegewässern findet oder die aus Plastikbehältern herausgelöst werden, auf das menschliche und tierische Hormonsystem Einfluss nehmen.
Über virtuelles Screening an der Universität Innsbruck wurden UV-Filter, die in Sonnencremes und Plastikmaterialien als Lichtschutz verwendet werden, in den Verdacht gebracht, die Testosteronbildung im Hoden zu hemmen. In-vitro-Studien an der Universität Basel bestätigten diese Befürchtung für einige Vertreter dieser Klasse, allen voran Benzophenon-1. Weitere Studien in vivo werden zeigen, ob es für die Verwendung dieser UV-Filter nicht strengere Regeln geben müsste.
Die Möglichkeiten von computerunterstütztem Wirkstoffdesign sind also nicht nur auf die klassische Arzneistoffentwicklung beschränkt, sie können auch wertvolle Hinweise zu Bioaktivitäten von Naturstoffen und Chemikalien liefern. Diese Methoden sind fester Bestandteil in der modernen Arzneimittelforschung, weil sie ein tieferes Verständnis für die molekularen Wirkmechanismen von Substanzen ermöglichen und so einen wertvollen Beitrag für eine effiziente Arzneistoffentwicklung leisten. Die fortschreitende Verbesserung ihrer Methoden und die vermehrte Zusammenarbeit zwischen Modeling-Gruppen und Experimentatoren werden in naher Zukunft schöne Früchte tragen.
Legende zu Abb. 1: Der Dopaminrezeptorantagonist Eticloprid in der Bindetasche des Dopamin-D3-Rezeptors (Koordinaten aus der Protein Data Bank, www.pdb.org, Eintrag 3PBL). Von den beobachteten Protein-Ligand-Wechselwirkungen (gelb: hydrophobe Wechselwirkung, blauer Stern: positiv geladene Wechselwirkung, Programm LigandScout von Inte:Ligand GmbH Wien) werden allgemeine chemische Eigenschaften von Dopaminrezeptorantagonisten z. B. in Form eines solchen Pharmakophormodells abgeleitet, welche die Basis für virtuelle Aktivitätsvorhersagen bilden.
Legende zu Abb. 2: Celecoxib in der Cyclooxygenase-2 Bindetasche (Protein Data Bank Eintrag 3LN1 dargestellt mit LigandScout). Der Hemmer könnte noch optimiert werden, indem man dem roten Pfeil folgend die derzeit leere hydrophobe Tasche (schwarzes Oval) ausfüllt, etwa durch zusätzliche Methyl- oder Ethylsubstituenten am 5-er Ring.






