
Gesetz zur Neuordnung des Arzneimittelmarktes (AMNOG): Was ändert sich?
15.06.2011 -
-
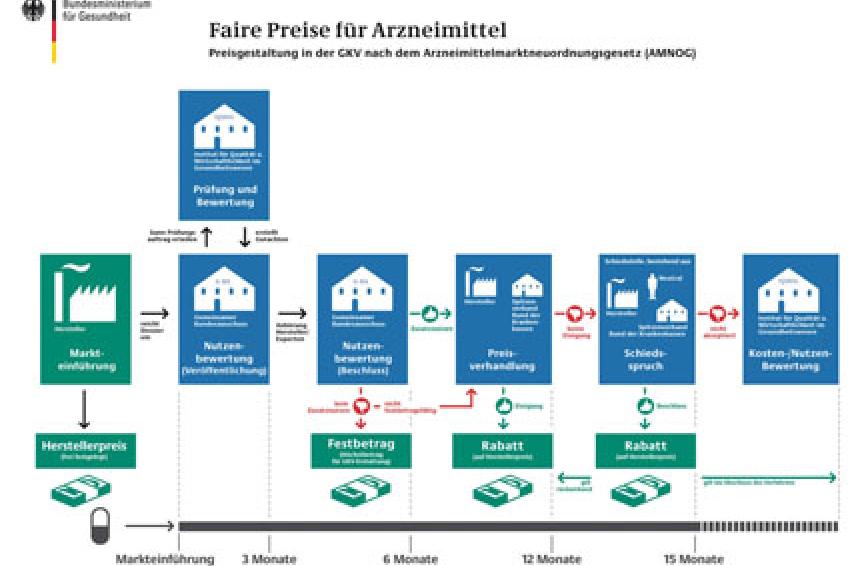 Abb. 2: Schematische Darstellung des AMNOG-Verfahrens (Quelle: Bundesministerium für Gesundheit).
Abb. 2: Schematische Darstellung des AMNOG-Verfahrens (Quelle: Bundesministerium für Gesundheit). -
 Abb. 4: Tabellarische Zusammenstellung der Zusatznutzen-Bewertung nach den Vorgaben des GBA.
Abb. 4: Tabellarische Zusammenstellung der Zusatznutzen-Bewertung nach den Vorgaben des GBA. -
 Abb. 3: Übersicht der Evidenzklassen für Klinische Untersuchungen.
Abb. 3: Übersicht der Evidenzklassen für Klinische Untersuchungen. -
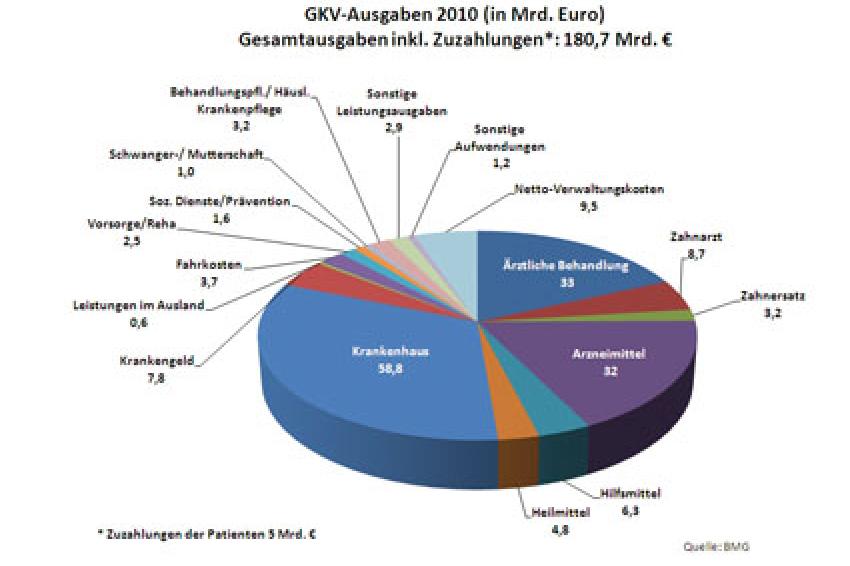 Abb. 1: Ausgaben in der GKV in 2010
Abb. 1: Ausgaben in der GKV in 2010 -
 Prof. Dr. Rainer Riedel, Institut für Medizinökonomie & Medizinische Versorgungsforschung (iMÖV), Rheinische Fachhochschule Köln
Prof. Dr. Rainer Riedel, Institut für Medizinökonomie & Medizinische Versorgungsforschung (iMÖV), Rheinische Fachhochschule Köln
Im Geltungsbereich der gesetzlichen Krankenkassen erhöhten sich die Gesundheitsausgaben in Deutschland von rd. 140 Mrd. € im Jahr 2004 auf rd. 175 Mrd. € im Jahr 2010.
Die Arzneimittelausgaben stiegen in diesem Zeitraum von 22,8 Mrd. € (2004) auf rd. 32 Mrd. € im Jahr 2010. Durch eine Reihe von Maßnahmen war man bestrebt, die Arzneimittelausgaben in den vergangenen Jahren „zu regulieren". Exemplarisch seien hier die Arzneimittelbudgets der niedergelassenen Ärzte, die Arzneimittel-Festbeträge und die Rabattverträge insbesondere für Wirkstoffgruppen aus dem Bereich des Generika-Segments genannt. Nach aktuellen Zahlen des BMG konnten die gesetzlichen Krankenkassen die erzielten Einsparungen durch den Abschluss von Arzneimittelrabatt¬verträgen von 846 Mio. € im Jahr 2009 auf rd. 1,1 Mrd. € im Jahr 2010 erhöhen.
Da es durch die oben aufgeführten Regelmechanismen nicht gelang, die Arzneimittelausgaben in Deutschland zu stabilisieren, entschloss man sich, politisch den Markt für innovative Arzneimittel in Zukunft zu regulieren. Im Rahmen dieses Entscheidungsprozesses wurde das nachstehende grundsätzliche Bewertungs- und das daraus resultierende „faire Preisfindungsverfahren für innovative Arzneimittel" durch das AMNOG (§ 35a SGB V) eingeführt. Der Verfahrensweg für die Preisfindung im Markt für innovative Arzneimittel ab dem 01.01.2011 wird nachfolgend kurz skizziert:
- Zulassung des innovativen Arzneimittels bei der zuständigen Zulassungsbehörde; hierfür war und ist es erforderlich, den Nachweis der Wirksamkeit und der Unbedenklichkeit (placebogestützte Studien) für dieses Arzneimittel zu erbringen.
- Mit Markteinführung (Anm.: nach der Zulassung) muss das pharmazeutische Unternehmen ein Nutzen-Dossier beim Gemeinsamen Bundesausschuss einreichen. Im Rahmen dieses Dossiers soll der therapeutische „Zusatznutzen" für das innovative Arzneimittel im Vergleich zu einer wirtschaftlichen Vergleichstherapie belegt werden. Der GBA hat am 28.01.2011 eine ausführliche „Handlungsanweisung" veröffentlicht. Auf dieser Grundlage haben die pharmazeutischen Unternehmen ihre Dossiers zu erarbeiten.
- Der GBA beauftragt dann z.B. das IQWiG als unabhängiges wissenschaftliches Institut, ein Bewertungsgutachten zu dem vorgelegten Dossier vorzunehmen. Eine abschließende Stellungnahme hinsichtlich dieser Nutzenbewertung für das innovative Arzneimittel muss innerhalb von drei Monaten durch das beauftragte Institut erarbeitet werden.
- Auf der Basis des „bewerteten Zusatznutzens für das innovative Arzneimittel" erfolgt eine Nutzenbewertung des GBA. Beim Vorliegen eines fehlenden Zusatznutzens wird dieses innovative Arzneimittel dann „einer Festbetragsgruppe zugeordnet.
- Liegt ein Zusatznutzen vor, kommt es im nächsten Schritt zu Preisverhandlungen zwischen dem Spitzenverband Bund der Krankenkassen (SpiBu) und dem pharmazeutischen Unternehmen. In Abhängigkeit des „bewerteten Zusatznutzens" sollen die Vertragsparteien einen „fairen Preis", unter Einbeziehung eines Rabatts auf den vom pharmazeutischen Hersteller zur Markteinführung gestellten „Arzneimittel-Preises", vereinbaren.
- Sofern sich die Vertragsparteien nicht auf einen „fairen Marktpreis" einigen können, wird der Sachverhalt der Schiedsstelle vorgelegt. Hier wird dann ein „Schiedsspruch hinsichtlich der zu erzielenden Preisfindung" erwirkt.
- Wird der Schiedsspruch von dem pharmazeutischen Unternehmen nicht akzeptiert, muss eine Kosten-Nutzen-Bewertung (KNB) durch das IQWiG erstellt werden. Bis zum Vorliegen der KNB-Ergebnisse gilt in diesem Fall zunächst der „Arzneimittelpreis" auf der Basis des Schiedsspruchs.
Die pharmazeutischen Unternehmen sind gehalten, den therapeutischen Zusatznutzen in klinischen Studien mit dem Evidenzgrad 1 a oder 1 b zu belegen. Zur Verdeutlichung der Studieneinteilung wird auf die Abb. 3 verwiesen
Um erfolgreich den Nachweis für einen bestehenden Zusatznutzen des innovativen Arzneimittels führen zu können, haben die pharmazeutischen Unternehmen eine Reihe von Herausforderungen zu bewältigen:
Welche adäquate wirtschaftliche Vergleichssubstanz ist in einer zu planenden Therapiestudie der Phase III für das innovative Arzneimittel zu wählen? (Anm.: Für zukünftige Studien kann sich das Unternehmen durch den GBA kostenpflichtig beraten lassen.)
Der Nachweis eines therapeutischen Zusatznutzens lässt sich i.d.R. nur durch eine „Superior-Arzneimittel-Studie" ¬belegen. (Anm.: Ein solcher Nachweis ist nachvollziehbarerweise für die in den kommenden Jahren zur Markt¬reife geführten innovativen Arzneimittel „nicht einfach zu führen", da diese neue „100 %ige-Reimbursement-Voraussetzung" bei den Studiendesigns vor fünf Jahren noch nicht bekannt waren.)
Es fehlen erfahrungsgemäß ausreichende Versorgungsforschungsdaten aus empirischen Feldstudien. Diese Daten sind jedoch erforderlich, um umfangreiche Kosten-Nutzen-Bewertungen vornehmen zu können.
Damit ein pharmazeutisches Unternehmen zukünftig in der Lage ist, einen „fairen Preis für sein innovatives Arzneimittel" zu erzielen, muss die „innovative Substanz" einen erheblichen Zusatznutzen nachweisen. Demgegenüber kann man mit einem angemessenen „Preiszuschlag" für die innovative Substanz auf dem Verhandlungswege dann rechnen, wenn für diese Substanz ein „bedeutsamer Zusatznutzen" nachgewiesen werden konnte.
In den kommenden Jahren wird somit voller Spannung die Frage zu beantworten sein, in welchem Umfang sich eine Nutzenverbesserung von Arzneimitteln mit einem entsprechend korrespondierenden Preiszuschlag gegenüber der wirtschaftlichen Vergleichstherapie auswirken wird.
Beispiel: Eine 15%ige Nutzenverbesserung wäre dann gleichzusetzen mit einem 15 %igen Preisaufschlag auf das Preisniveau der wirtschaftlichen Vergleichstherapie?
Hier wird es für alle Beteiligten aus den Bereichen der gesetzlichen Krankenkassen, dem IQWiG, dem GBA, der pharmazeutischen Industrie, der PKV, den Apothekern und den Patienten von großem Interesse sein, wie sich in den kommenden Jahren das Preisniveau für innovative Arzneimittel in Deutschland entwickelt? Aufgrund der zu erwartenden AMNOG-Einflüsse auf den deutschen Arzneimittelmarkt bleibt es abzuwarten, in welchem Maße die Zahl der innovativen Neuzulassungen in den kommenden Jahren zurückgehen wird oder nicht.
Kontakt
Rheinische Fachhochschule Köln
Schaevenstr. 1 a/b
50676 Köln
+49 221 20302 0
+49 221 20302 45








